Sys2Diag CNRS UMR9005. Cap delta/Parc euromédecine, F-34184 Montpellier cedex 4
LRI, Univ. Paris Sud, CNRS UMR8623. Bat 650, F-91405
Orsay cedex.
Abstract
|
Many of the biological objects (genetic regulatory networks,
metabolic networks, signalling pathways, etc.) biologists and modellers are
interested in, do not need to take into account the spatial location of the
biomolecules that are involved in the studied processes. In the cases, where
there are large populations of molecules, even a stochastic approach is not
absolutely necessary, and a deterministic method, such as ordinary differential
equations, gives useful results.
To study these kind of biological processes we need a well founded framework that can both account for the stochasticity and the low numbers of participants, but also for the spatial localisation of molecules. Finally, its implementation must be efficient in terms of computation time. We will use in this workshop the HSIM simulator which implements an hybrid simulation technology, mixing entity-centered modelling with a new approximated Stochastic Simulation Algorithm (SSA). |
The simulator, HSIM, is a stochastic automaton driven by reaction rules between molecules.
In essence, each molecule is represented by a record that includes its type, its position, its size and a list of links to certain other molecules. HSIM keeps track of each molecule in real time from the computer point of view. The basic principle is that time is sliced into consecutive steps or generations, and in each generation the rules are applied to every molecule. These rules mimic the chemical reactions between molecules in a real system. The generation time is set to 100 microseconds, which corresponds to the average time for a protein to move a distance of 10 nanometers (of the order of its diameter) in vivo.
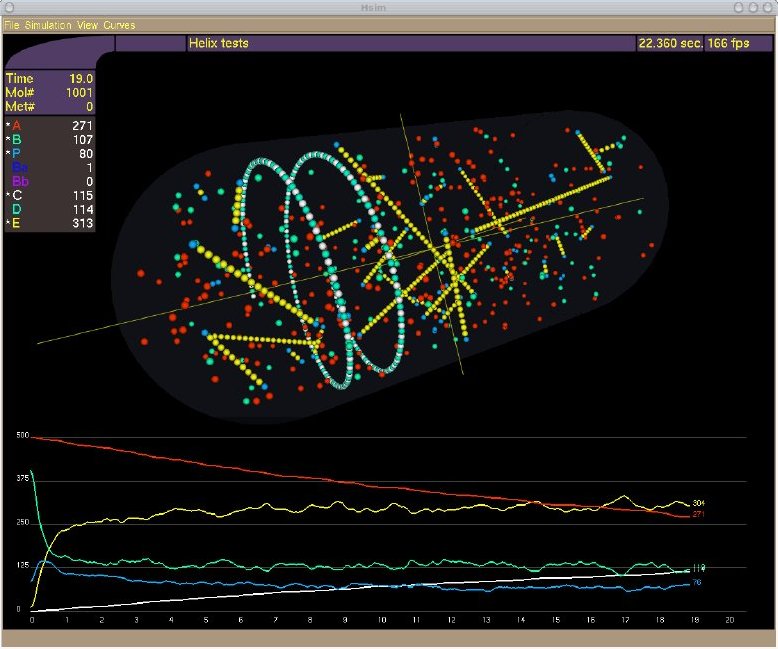
Metabolites diffuse faster than proteins, to take account of their smaller size, they are represented in HSIM by a sphere of reduced size with a greater diffusion speed.
Algorithm
During a generation, the following processes are applied to all the molecules:
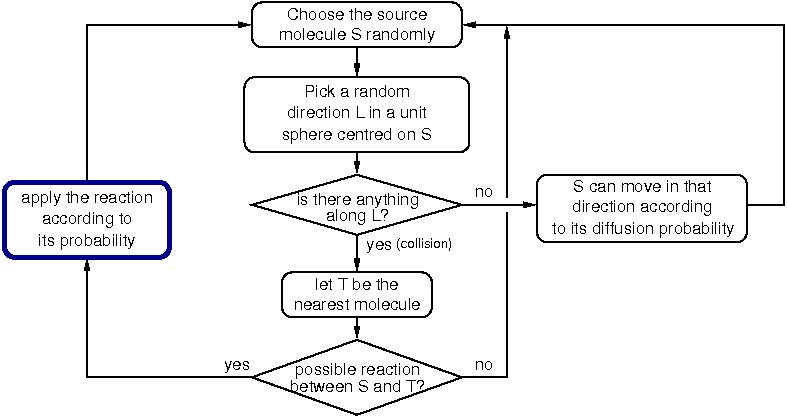
When all the molecules in the cell have been processed, the generation is completed and a new one begins. In HSIM the computer time is proportional to the total number of molecules and not to the size of the simulated space or the number of types of molecules.
One important point is that models in HSIM are additive: different models can be merged by simply merging their configuration files. If there are interactions between the models, HSIM will take them into account. Rules There are four kinds of interaction rules in HSIM between two molecules:
Each rule has an associated probability which corresponds to the kinetics of the reaction. For each kind of molecule, the maximum number of links to each other kind of molecule must be specified to allow the association rules to be functional. Model Description The model is described in a configuration file made of 5 sections:
Example
title = "Enzymatic Reaction";
geometry = 120:40; // 1.2 x 0.4 nm
molecule s1, s2;
molecule E1;
size (s1) = 0.1;
size (s2) = 0.1;
speed (E1) = 0.1;
maxlinks (E1) = s1(1), s2(1);
maxlinks (s1) = E1(1);
maxlinks (s2) = E1(1);
E1 + s1 -> E1 * s1 [0.4]; // E1 captures its substrate
E1 * s1 -> E1 + s1 [1e-3]; // reverse reaction
E1 * s1 -> E1 * s2 [0.01]; // E1 catalyses s1 -> s2
E1 * s2 -> E1 + s2 [0.01]; // E1 releases the product
init (30, E1); // 30 copies of E1
init (1000, s1); // 1000 copies of s1
Command line options
Usage: glhsim -f config-file [options]
-h print this help.
-H longer help (with interactive controls).
-b file batch mode (no OpenGL display).
-bd file batch mode (without diffusion phase).
-C file count each reaction and write it in 'file'.
-m num set the duration of the simulation (number of seconds of simulated time).
-q quiet (no display at all).
-v prints the rules on stderr.
-r num initialise the random number generator.
-R display the rules.
-fs display in full screen mode.
-s 3D stereo mode.
-f file use 'file' as configuration file.
-l file load the simulation snapshot 'file' (infers the configuration).
-w reload periodically the snapshot (watch file).
-g WxH set the cell width and height.
-i num set the number of generations between two histograms display.
-c MOL=num add to the initial population of MOL 'num' more copies.
Keyboard controls
a show all the molecules (even those not linked)
b show the backbone of the assemblies
d toggle diffusion only / diffusion and reaction
D set the length of the simulation in seconds
g toggle concentration curves / assemblies histogram
h, ? show this help
i save the current display in a PNG image file
l load a previously saved simulation
m start / stop recording a movie of the simulation
n normalise the scale for displaying the concentration curves
+ increase the scale factor
- decrease the scale factor
q, Escape exit the program
r show / hide the links between bound molecules
R show / hide the rules
s toggle the 3D stereoscopic mode switch
S save the current state of the simulator into a file
Tab start/stop the simulation
Return toggle display rate
Backspace focus to the center of the cell
Mouse controls
Left Drag rotate around the X and Y axis
RIght Drag change the aperture angle
Left Press select a molecule to be the new center of rotation
Ctrl+Left Press select an assembly to be shown
Mid Press show a menu
Model description language
- General syntax
title = "model name"; name of the model
speed (mt) = prob; diffusion speed expressed as a probability
size (mt) = num; diameter of a molecule type in 10 mn unit.
geometry = lengh:diameter; size of the cell in 10 mn units.
display (mt1, ..., mtN); show the concentration curves of the species list.
asm name = (mt1, ..., mtN); give the name name to all the assemblies containing the species list.
maxlinks (mt) = mt1 (nl1), ..., mtn (nln); set the maximum number of links for species mt.
molecule descr1, ..., descrn declare the species descr1, ..., descrn as cytosolic molecules where descri is mt~~[max link count] [hide] [inactive]
membrane descr1, ..., descrn declare the species descr1, ..., descrn as membrane molecules
metabolite mt1, ..., mtn declare the species as cytosolic molecules treated as an homogeneous population
init (#copies, mt); fill the compartment with #copies copies of species mt.
init (conc uM, mt); fill the compartment with conc micromolar of species mt.
init (conc mM, mt); fill the compartment with conc millimolar of species mt.
surface (#copies, mt); put #copies copies of the membrane species mt on one pole of the compartment membrane.
Syntax of the reaction rules
- Basic reactions
Each molecule type of the left side of a rule can be more specific than simply the species name.
The binding context can be expressed with this syntax:
mt1 + mt2 -> mt3 + mt4 [prob];
mt1 reacts with mt2 with probability prob;
mt1 + mt2 -> mt3 * mt4 [prob];
mt1 reacts with mt2 with probability prob and forms a complex where mt1 become mt3 and mt2 become mt4.
mt1 * mt2 -> mt3 + mt4 [prob];
the complex mt1 / mt2 dissociates with probability prob and mt1 become mt3 and mt2 become mt4.
mt1 * mt2 -> mt3 * mt4 [prob];
the complex mt1 / mt2 reacts with probability prob to transform mt1 to mt3 and mt2 to mt4.
mt
an instance of molecule type mt, bound or not to any other molecule
{mt1}mt
an instance of molecule type mt which is already bound to a instance of molecule type mt1
{~mt1}mt
an instance of molecule type mt which is not bound to a instance of molecule type mt1
- Enzymatic reactions
A specific syntax has been implemented to model enzymatic reactions, allowing
to specify the kinetics with the usual constants Km and Kcat, and units
µM and mM. For example:
geometry = 60:60;
molecule GOD; // Glucose oxydase. Km = 30 mM, Kcat = 337
metabolite glucose, h2o2;
GOD (gluc -> h2o2) Km = 30 mM; Kcat = 337;
To implement this kind of reaction, HSIM use 3 standard rules and compute their
probabilities to match the Km and Kcat values:
GOD + gluc -> GODgl [0.04884]
GODgl -> GOD + gluc [0.8]
GODgl -> GOD + h2o2 [0.0674]